Artificial intelligence in molecular design
Jody Dascalu | February 20, 2023In the past few years, artificial intelligence (AI) has evolved from theoretical studies and research to real-world applications. Advances in AI and machine learning (ML) algorithms such as deep learning (DL) have allowed machines to imitate human intelligence by learning from data. Consequently, AI and ML approaches may revolutionize molecular design.
Prior to AI, when experimental laboratories designed new compounds, they relied almost completely on experts to design experiments and characterize, validate and analyze final products. This bottleneck has slowed the discovery of new materials, which could have implications for many industries, such as electronics, drug discovery and energy storage.
 Photomicrographs of the drug AZT. Source: NIH Image Galery/CC BY-NC 2.0
Photomicrographs of the drug AZT. Source: NIH Image Galery/CC BY-NC 2.0
The many materials in use today are the result of exploring only a small portion of the theoretical chemical space. Undiscovered materials may bring unprecedented advances to technologies. In the past, automated molecule discovery was met with limited success. With further developments in computing architecture, characterization techniques, and with material synthesis, DL-based models have helped this topic re-emerge as an approach of potential interest.
Molecular representations in AI molecule discovery
Computers process data in bits and cannot interpret images in the same way a person can. Data that the computer interacts with needs to be converted into a format that the computer is able to process and “understand.” The way molecules are represented can influence AI-driven drug discovery.
 Examples of molecular representations. Source: Vladsinger/CC BY-SA 3.0
Examples of molecular representations. Source: Vladsinger/CC BY-SA 3.0
The advent of computers led to machine-readable chemical representations. With computers, compounds and their structures could be rapidly queried and stored. Algorithms help visualize compounds as 2D depictions and the visualization of compounds in 3D was also popularized. Skeletal structures of molecules are often referred to as 2D depictions.
Many neural networks learn a vector representation for molecules in the training data set and use that learned representation to predict molecule properties. Optical character recognition systems rely on ML and pattern recognition techniques to translate 2D chemical compounds to standard chemical representations.
With the development of graph neural networks, the use of molecular graph representation for property predictions and de novo design has advanced. De novo design uses incremental construction on a structure and ligand model. It is a methodology based on information regarding the biological target or active binders. Prior to this, compact linear notations such as SMILEs strings have been favored in ML applications as molecular graph representations have larger memory requirements.
In protein structure prediction using AI, multiple representations might be used. For example, a protein sequence could be the initial starting point and then a 3D model of the structure can be generated. Advanced molecular dynamic methods can be used to estimate how the protein folds and what the final structure of the protein could be.
Having an accurate picture of proteins and how they interact in a given disease helps scientists and AI to develop molecules for the correct targets. Researchers such as Pillong and Schenider applied their pseudo-receptor model in a virtual screening study to identify aminoglycoside scaffold replacements with antibacterial potential. ML can be used to investigate the interactions between glycans and proteins.
AI-driven discovery and design
Creating new chemicals and materials relies on molecular design. Traditional processes involved trial and error, guesswork and sometimes a little bit of luck. First, the chemical structure of the new target substance needs to be selected after careful evaluation of its expected properties. Next, designing the molecule requires finding chemical reactions that link up to create the target molecules or a combination of molecules. Synthesis plans can have hundreds of steps and create undesired by-products or might just not work at all. The process can be time-consuming and generate waste.
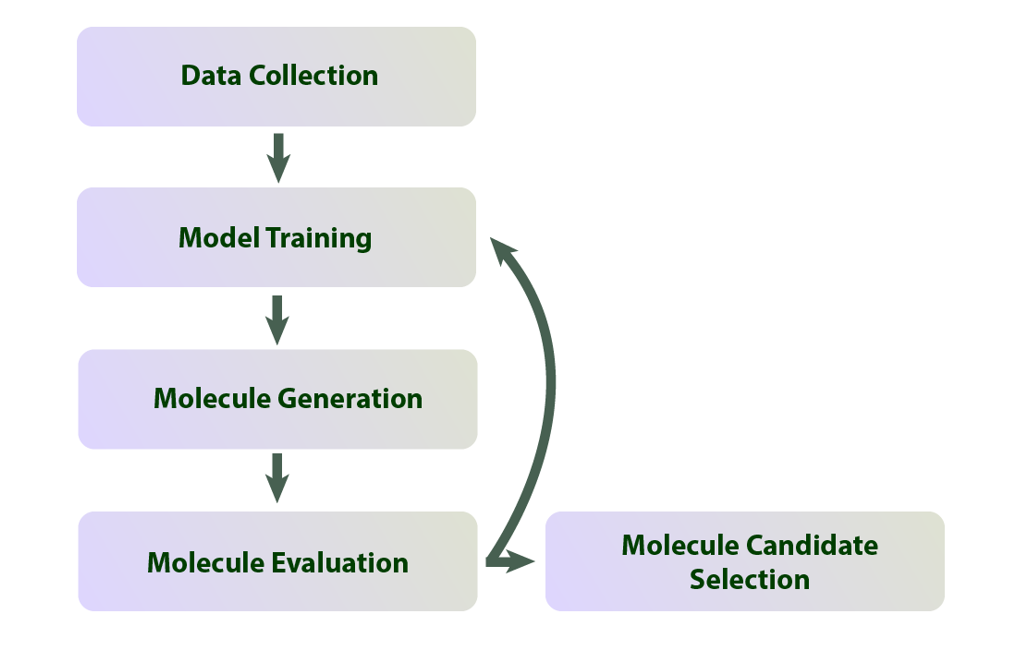 An example process for AI-driven molecular design. Source: Jody Dascalu
An example process for AI-driven molecular design. Source: Jody Dascalu
Some researchers estimate that there are between 1020 and 1024 drug-like organic compounds. When AI uses a data set of known chemical structures aligned with their properties, it can construct new molecules that have similar and maybe even more useful characteristics. AI can be used in this way to accelerate molecular design and drug discovery.
AI uses ML algorithms that can analyze data from past experiments and predict the structure of new molecules and generate manufacturing processes. In molecular design, specifically generative ML may play an important role in the commodification of AI in this application. DL generative models show promise as a way to move out from predetermined rules based on traditional knowledge-based or reaction rules.
A major problem in drug discovery is the complexity of human biology. Finding therapeutic agents that can target biological processes effectively while having little to no impact on other processes can be difficult. However, novel systems-based approaches can help speed up early-stage drug discovery. Cheminformatics and bioinformatics lead to faster and more efficient drug discovery.
AI approaches gaining momentum
Many organizations are funding research on AI in molecular design. The U.S. government has funded millions for the Materials Genome Initiative, which uses computational methods along with AI to establish an infrastructure that accelerates the development of materials.
An effective way to lower costs in the pharmaceutical industry would be to improve the success rate of clinical trials. The probability of approval for drugs entering clinical trials was around 10% in 2015 to 2017. It is estimated that around 80% fail because the candidate drug does not meet safety and efficacy criteria for patients. AI can help estimate drug efficacy and interactions with human biology, which could increase the drug approval rate.
New chemical compounds can pose unforeseen risks, but AI can be used to anticipate and reduce undesirable outcomes. DL models can also be used to predict drug effectiveness. For example, CDRscan was able to identify approved oncological and nononcological drugs that had potential cancer indications.
Drug repurposing strategies focus on using failed or abandoned compounds for different applications. These strategies rely on factors such as the association between drugs, diseases and targets, and ML can be used in drug repositioning. DL that focuses on using chemical information from drug repurposing can also be used.
In closing
Selecting compounds that have appropriate pharmacokinetic properties and create the appropriate cellular and physiological responses is vital. Screening compound libraries using a variety of techniques is often the earliest stage of drug design. Typically, compounds that have promising potency and absorption, along with distribution and toxicity, are chosen as lead compounds that need to be optimized. Many datasets have been gathered, but often disproportionately focus on a comparatively small range of well-studied endpoints.
Physics-informed ML has helped advance molecular design algorithms. As well, high-end hardware development has increased processing power. Using quantum, physics-based molecular representations and data generation tools are promising for a more widespread application of AI molecular discovery systems.



