Machine Learning Predicts Material Faults
Engineering360 News Desk | March 09, 2017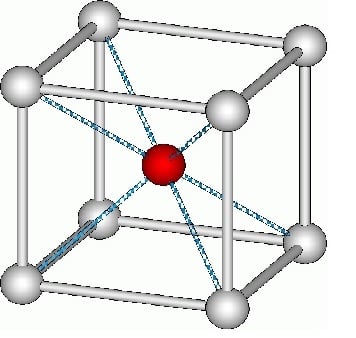 Intermetallic (B2) crystal structure Credit: Rene Töpfer/GNU FDLMachine learning algorithms developed by Lawrence Berkeley National Laboratory (Berkeley Lab) researchers can predict defect behavior in certain intermetallic compounds with high accuracy. This method will accelerate research of new advanced alloys and lightweight new materials for applications spanning automotive to aerospace more.
Intermetallic (B2) crystal structure Credit: Rene Töpfer/GNU FDLMachine learning algorithms developed by Lawrence Berkeley National Laboratory (Berkeley Lab) researchers can predict defect behavior in certain intermetallic compounds with high accuracy. This method will accelerate research of new advanced alloys and lightweight new materials for applications spanning automotive to aerospace more.
The new algorithms provide an alternative to the density functional calculation method of defect prediction. The existing method is computationally very expensive to use, limiting the scope of this testing.
The Berkeley Lab team selected 100 intermetallic compounds from the Materials Project Database, focusing on the common B2 crystal structure, and ran density functional calculations on them. Using this small sample of materials, the team used a forest machine learning model called gradient boosting to achieve a highly-accurate structure predictions.
In this approach additional machine learning models were built successively and combined with prior models to minimize the difference between the models predictions and the results from density functional calculations. The researchers repeated the process until they achieved a high level of accuracy in their predictions.
The new machine learning algorithms are both much less expensive to use and faster to run. These advantages should enable faster materials design. An extension of this work resulted in an open source Python toolkit (PyCDT) for modeling point defects in semiconductors and insulators.